Introduction
There are many innocuous retinal findings which arise from normal age-related change. It is important to distinguish sclerochoroidal calcifications (SCC) as either benign or malignant. Sclerochoroidal calcifications are pale, elevated fundus lesions found in the sclera, choroid, and/ or retina that originate from excess calcium deposition onto normal tissue. This uncommon finding is often benign, and the patient suffers no visual or systemic complications. However, in rare circumstances, these retinal lesions can originate from potentially harmful systemic conditions. A careful ophthalmic and systemic work up is needed to determine the lesion’s etiology and if intervention is required. This case demonstrates a causal relationship between a SCC fundus lesion and primary parathyroid cancer.
Case Presentation
A 62-year-old Caucasian male presented to the optometry clinic for a comprehensive eye exam. The patient had no ocular or visual complaints. His past ocular history included cataract extraction OU and mild hyperopia OU. Pertinent medical history consisted of multisystem vascular disease including IDDM type 2 for six years, hypertension, and hyperlipidemia.
The patient’s presenting visual acuities were 20/25 sc OD, OS, OU. Intraocular pressures, extra ocular motilities, confrontation fields, and pupillary testing were all normal. Slit lamp examination was unremarkable and revealed no calcium deposition on the cornea or conjunctiva.
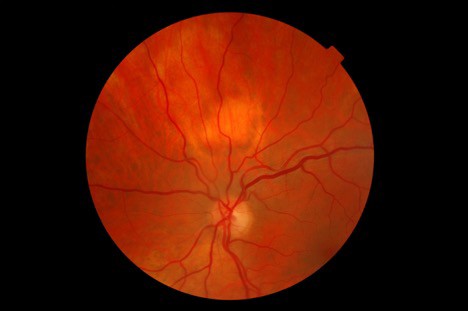
Dilated fundus exam revealed a 2DD pale, elevated lesion 1.5 disc diameters superior to the optic nerve head OS. (Image 1). Posterior segment evaluation was otherwise normal in both eyes. There were no concurrent lesions in the fellow eye. There was no evidence of diabetic or hypertensive retinopathy in either eye. The intraocular lens implants were well centered and clear in both eyes. The vitreous was also clear in both eyes.

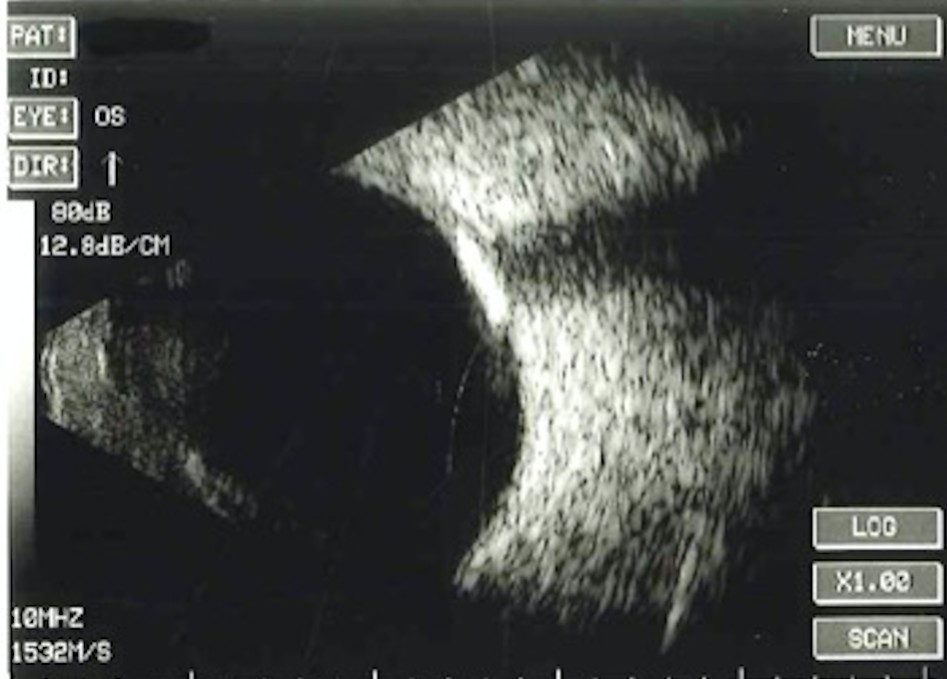
B-scan ultrasonography was performed on the lesion. The test confirmed a dense hyper- reflective area with acoustic shadowing. The size of the lesion was 3.1mm vertically and horizontally. (Image 2).

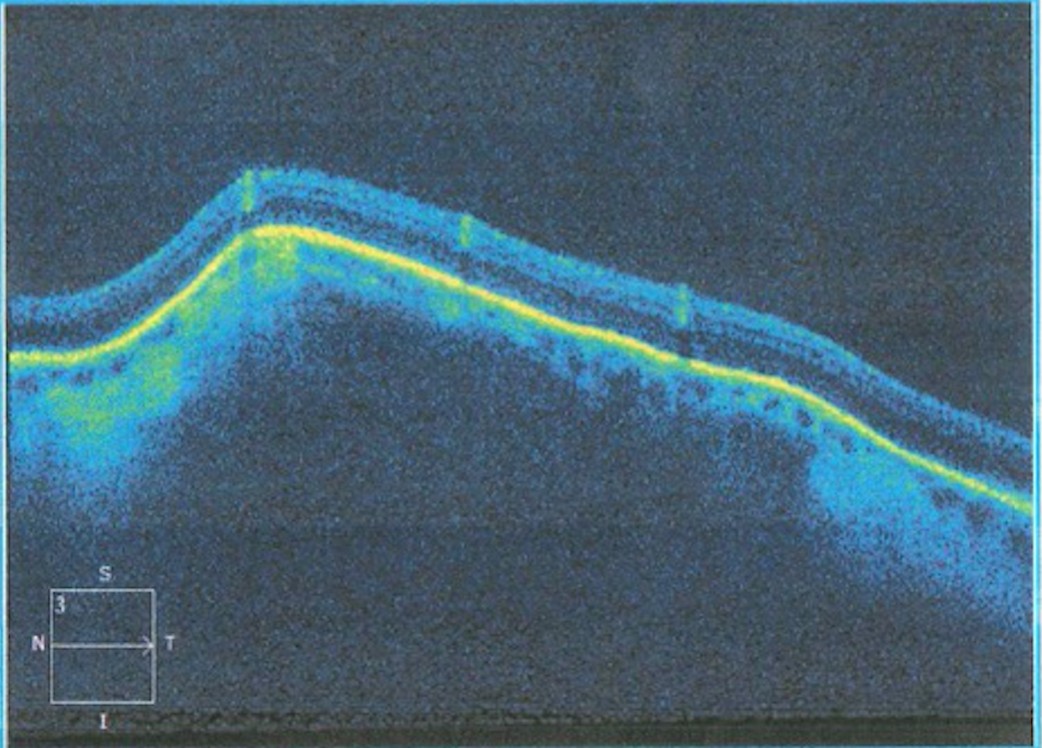
Optical coherence tomography with enhanced-depth imaging was then performed (OCT-EDI). There was steep elevation of the sclerochoroidal region underlying an intact retina. (Image 3). No neovascular membranes or vulnerabilities in the Bruch’s complex or ellipsoid zone were visualized.

Following presentation in the eye clinic, a physical, urinalysis, and metabolic work up were ordered. Laboratory testing included complete blood count, parathyroid hormone, calcitonin, calcium, magnesium, phosphorous, potassium, and alkaline phosphate levels.
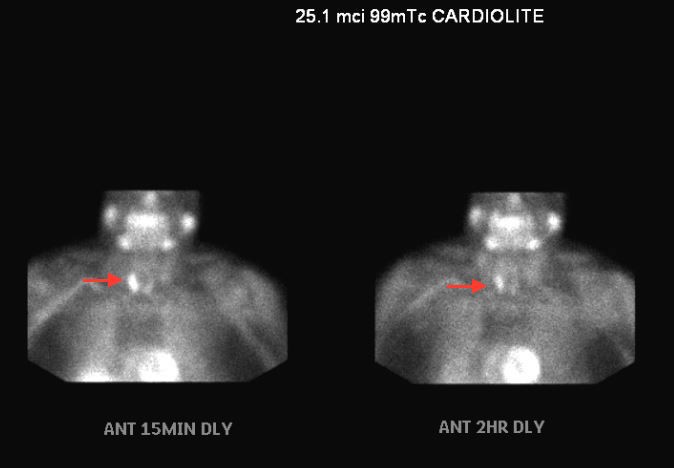
Parathyroid hormone was significantly elevated to 124.9pg/ml (normal range 29-79 pg/ml). Calcium levels were also mildly elevated to 10.3 mg/dl on two occasions (normal range 8.4-10.0 mg/dl). Due to abnormal metabolic markers and correlative ophthalmic findings, the patient was referred to endocrinology and oncology where more imaging was conducted. Parathyroid immunoassay with radioactive 99mTc-MIBI was positive for abnormal uptake on the right parathyroid gland side. (Image 4).

In multiple projections, the parathyroid region demonstrated focal uptake of radiotracer activity of the inferior aspect of the right parathyroid gland. There was washout of radiotracer activity throughout the left gland. Abnormal uptake was present on both fifteen minute and two-hour delay assessment. The patient was therefore diagnosed with primary hyperparathyroidism secondary to a right-side parathyroid adenoma. The patient is currently being considered for surgical resection of the parathyroid and subsequent radiation.
The ocular and visual complications of sclerochoroidal calcification are minimal. Vision is typically stable and unaffected. It is possible albeit rare to develop lesion enlargement, serous detachment, choroidal neovascular membranes and deposition of conjunctival and corneal calcium. Therefore, the patient is recommended to undergo routine dilated fundus examinations every six months and then annually. The overall visual prognosis is optimal. Fortunately, the systemic prognosis is guarded but hopeful: the 5-year relative survival rate for primary parathyroid cancer is 93% and the 10-year survival rate is 82%.1
Discussion
Metastatic calcification is a process describing inappropriate calcium deposition in normal tissue throughout the body. Sclerochoroidal calcification pertains to metastatic calcium deposition primarily involving the normal tissue of the sclera, choroid, and rarely the retina. It is believed that the sclera becomes involved first, often leaving the fundus appearance unchanged.2 Progression of the condition is indicated by choroidal involvement and atrophy of the retinal pigmented epithelium as well as other retinal layers. In very advanced cases, focal areas of serous and hemorrhagic retinal detachments and choroidal neovascular membranes may ensue due to extensive chorioretinal degeneration. These are very rare complications, in fact only three cases have been reported in literature to date.3 Total retinal detachment has not been documented due to SCC.
Most recent clinical evidence suggests that SCC lesions do not change either by progressing or resolving despite systemic intervention to treat the underlying pathology. A 2001 study conducted by Honovar et al analyzed the demographics of 27 patients and 38 eyes diagnosed with SCC lesions over 38 months. All patients continued to remain asymptomatic, with stable visual acuity, stable lesions, and no new lesions. Only 1 of 27 patients was had evidence of primary hyperparathyroidism."4 The Shields et al study of 179 eyes concluded similarly at an average of 4 years follow up, there was no lesion enlargement, decalcification, or related subretinal fluid/hemorrhage, choroidal neovascularization, or vision loss. Ocular intervention or treatment was not necessary in any case, and systemic associations linked SCC to hyperparathyroidism (27%) with parathyroid adenoma (15%), Bartter syndrome (2%), or Gitelman syndrome (11%).5
Systemic implications can vary although most SCC lesions are idiopathic. In a very small number of cases, SCC is the result of primary parathyroid cancer, a potentially fatal condition.
The parathyroid gland consists of cells that synthesize and secrete parathyroid hormone which regulates calcium and phosphate metabolism. An abnormal proliferation of parathyroid cells due to either a benign or metastatic tumor increases calcium efflux from bones, where it deposits inappropriately in other tissues such as the eye, skull, phalanges.6,7 Abnormalities of calcium levels in the body can have a vast array of complications from brittle and painful bones to neurological changes. In other cases, SCC can result from Bartter and Gitelman Syndromes, which are autosomal recessive anomalies of the sodium and chloride transport system of cells. Sodium/calcium transport issues result in hypokalemia and metabolic alkalosis.2 Patients who suffer from these conditions have chromosomal defects which cause elevated levels of excreted electrolytes in urine and other systemic signs.
Sclerochoroidal calcifications can simulate many intraocular growths including choroidal melanomas, choroidal osteomas, choroidal metastases, intraocular lymphomas, or choroidal nevi. In a study conducted at the Oncology Service at Wills Eye Hospital, practitioners incorrectly identified referred SCC lesions as choroidal metastasis (26%), choroidal melanoma (21%), choroidal nevus (11%), intraocular lymphoma (3%), and unknown lesion (39%).2
While it is important to correctly diagnose all sclerochoroidal calcifications, an important distinction should be made between sclerochoroidal calcifications and choroidal osteomas. They have the most similar pathophysiology but the visual and systemic prognosis is different for each. The visual prognosis for SCC is very good, while osteomas may leave patients visually impaired. Osteomas present with progressive vision loss that plateaus around 20/400.
Systemically, SCC has the potential for fatal systemic complications, as in the case above. Osteomas are not related to systemic complications and are isolated to the ophthalmic region. Epidemiology can be helpful when differentiating clinical appearance. Both SCC and osteomas can mimic pale-yellow, elevated lesions. However, sclerochoroidal calcifications are reported bilateral in 46% of cases and prominently located in the superotemporal quadrant (85% of the time) between the temporal arcades and the equator.8 Osteomas are more often to be unilateral, solitary masses which are juxtapapillary or parapapillary. SCC are common in older males, and osteomas are found in younger females between the second and third decade of life.
Hasanreisogl, M. et al. proposed a classification system of four distinct types of SCC lesions which can be distinguished primarily by EDI-OCT, although B-scan and fundus appearance aid in the distinction. The types and characteristics are as follows. Type 1 is characterized by a completely “flat” lesion which is only visible by focal thinning of the choroid on EDI-OCT. Type 2 has a “rolling” appearance which is evident on stereoscopic views of the fundus as well as thinning of the choroid overlying the apex of the lesion on EDI-OCT. Type 3 is characterized by a “rocky-rolling” appearance on fundoscopy. Type 3 also has clear thinning of the overlying choroid as well as thinning and at times obliteration of the outer nuclear layer (ONL) and external limiting membrane (ELM). Type 4 has a “table-mountain” appearance and a well-preserved choroid with no areas of thinning overlying the top of the lesion.8
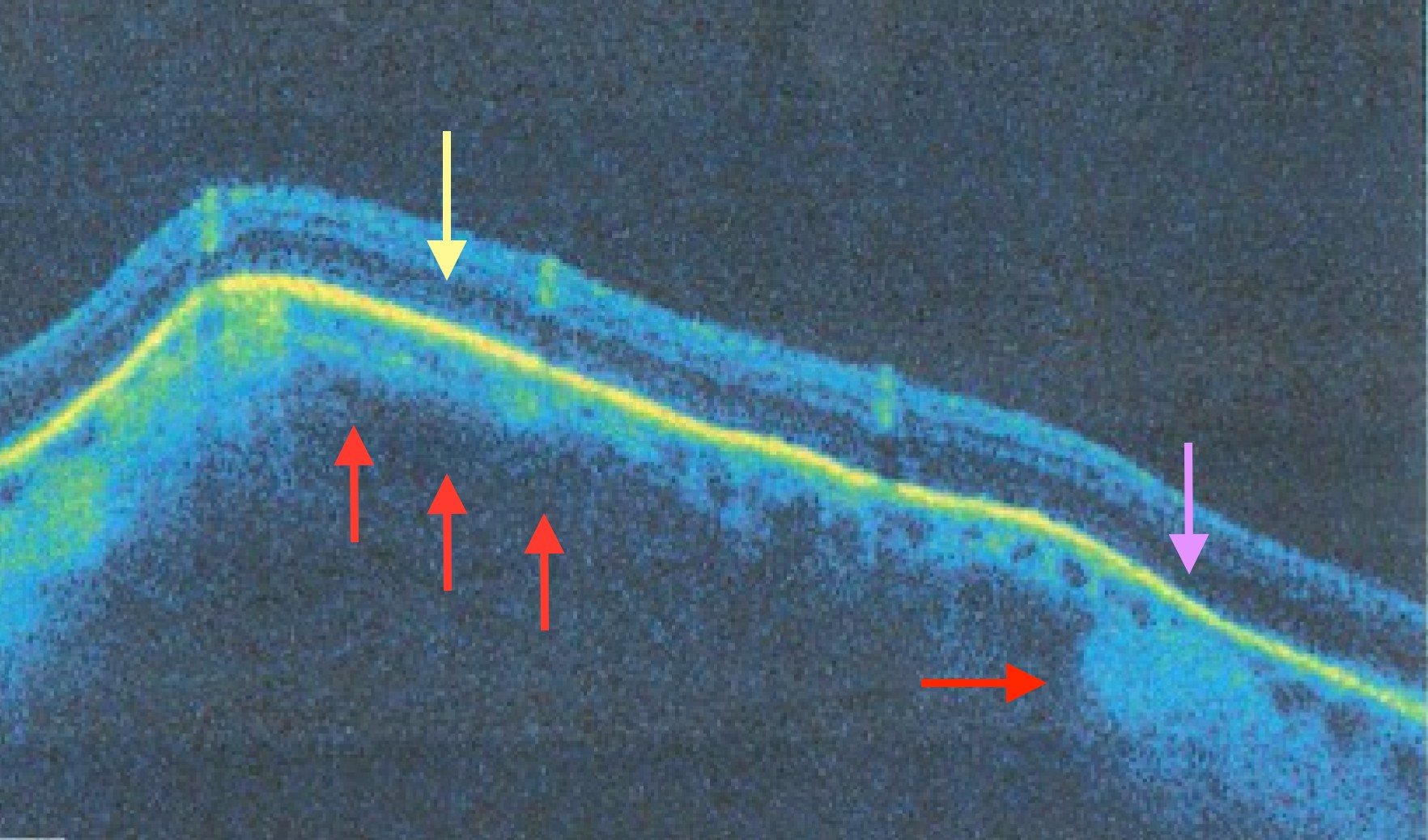
The patient described above was diagnosed with a Type 3 sclerochoroidal calcific lesion. (Image 5). The image illustrates red arrows pointing to a thinned choroid towards the apex of the lesion compared to the thicker choroid at the temporal aspect of the lesion (on the right side of the image). The yellow arrow points to an area with thinned outer nuclear layer, and the purple arrow points to an absent external limiting membrane. The type of lesion is not yet clinically specific of pathophysiology and cannot be used to distinguish benign and malignant fundus lesions.

Conclusion
Sclerochoroidal calcification should be considered as a differential diagnosis when examining patients with elevated fundus lesions. Fundoscopic clinical appearance should be compared to systemic review to determine if further work up is required. At minimum, patients with newly presenting SCC lesions should be referred for physical and complete blood work including calcium and parathyroid function. Any systemic anomalies should be thoroughly vetted and correlated with both systemic and fundoscopic clinical findings. Consultation with endocrinology for high-risk cases is advised for best continuity for care.
Further study into the characteristics differentiating the types and presentations of SCC lesions as analyzed by OCT and B-scan may help clinicians better understand the pathophysiology and therefore best treatment practices for patients in future.
Conflict of Interest and Financial Disclosure
The authors declare that there are no conflicts of interest and no financial ties relevant to the topic at hand.