Introduction
Ectopia lentis is a partial displacement of the intra-ocular lens. There are a number of causative factors for ectopia lentis, which can be classified as familial or secondary to trauma.1 Marfan syndrome, one of the most commonly inherited connective tissue disorders, can cause ectopia lentis. The pathogenesis of Marfan syndrome results from an inherited defect in the extracellular glycoprotein fibrillin-1.2–10 Marfan syndrome can affect the skeletal, ocular, and cardiovascular systems. Marfan syndrome can lead to an aortic dissection and aneurysm, causing serious illness or death.
Case Presentation
A 30-year-old Caucasian male (JA) presented to the refractive surgery clinic for a refractive surgery screening. JA only reported mild blur at distance with each eye when not wearing any glasses for the past year. JA previously had PRK in 2006 and reported that his vision was good until a year ago. JA reported a long history of cardiovascular problems including pericarditis, for which he was currently taking colchicine. JA reported that the cause of his recurrent pericarditis was still unknown, but he was currently under the care of a cardiologist. When questioned further about the pericarditis, he reported that it began during a deployment in the Middle East a few years ago. He was medically evacuated back to the U.S. due to the diagnosis. JA reported that he had a trans-thoracic echo cardiogram earlier in the spring which did not reveal any reason for the pericarditis. Other medical history revealed that the patient’s grandmother had glaucoma and the patient had an allergy to penicillin. JA was oriented to person/place/time.
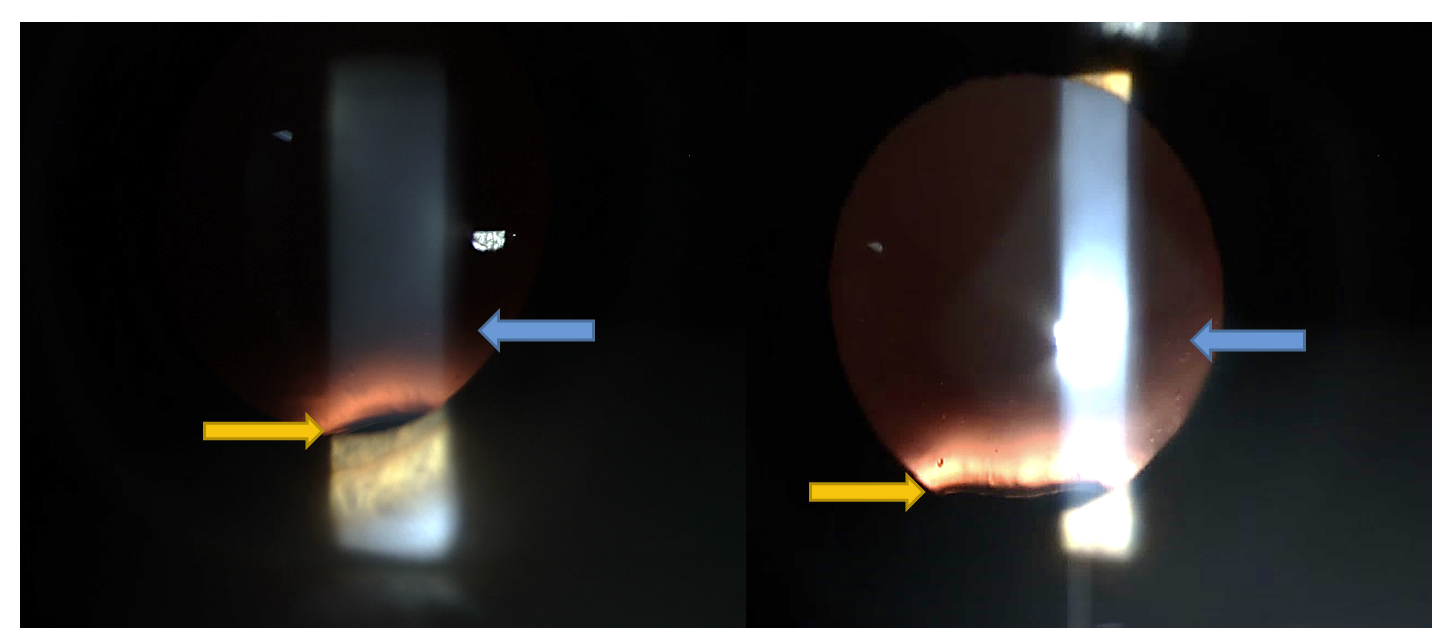
JA’s uncorrected visual acuity was 20/25- at distance and 20/20 at near with the right eye and 20/40+1 at distance and 20/20 at near with the left eye. Best corrected visual acuity was 20/15 with the right eye and 20/15 with the left eye with a manifest refraction of -1.00-1.25x015 OD and -2.00-1.25x175 OS. Preliminary testing was all within normal limits, with no strabismus or phoria detected on cover test, full range of movement on extra-ocular muscle testing, and normal pupil responses. Both pupils measured 5 mm in dim light and 4 mm in bright light. Confrontation visual field testing was full and normal with each eye. Intra-ocular pressures were 16 mm Hg OD and 15 mm Hg OS via non-contact tonometry. Anterior segment evaluation via slit lamp examination was within normal limits with no pathology observed. JA was dilated with one drop of 2.5% phenylephrine and one drop of 1% cyclopentolate in each eye. An evaluation of the posterior segment was performed with a 78D lens and BIO/20D lens. Examination of the intra-ocular lenses immediately revealed superior subluxation of both lenses as seen in Figure I. The vitreous was clear bilaterally, with no evidence of separation. The optic nerves appeared normal with healthy color, normal nerve fiber layer, and a cup-to-disc ratio of .2 in both eyes. The posterior pole and peripheral retina were flat and intact with no pathology observed. JA was educated about the subluxed lens findings and that he should not undergo another refractive surgery procedure at this time.

The primary differential diagnoses considered in this case of ectopia lentis included:
-
Homocystinuria
-
Marfan syndrome
-
Weill-Marchesani Syndrome
-
Sulfite Oxidase Deficiency
-
Trauma
A panel of lab tests was ordered to explore the cause of the patient’s ectopia lentis. The patient was referred back to his cardiologist for further testing. The cardiologist decided to perform a CT angiogram to assess for vascular abnormalities and, specifically, an aortic dissection. A genetic test was also ordered to look for any abnormalities of the FBN1 gene, which could be indicative of Marfan syndrome.
The RPR lab was negative. Cardiology perfomed the CT pulmonary angiogram. The angiogram revealed that the aorta was of normal caliber without dissection and no pleural effusion was detected. The urine and plasma amino acid analysis did not show a pattern that would suggest a specific inherited metabolic defect. Both the methionine and homocysteine levels were normal in the plasma and urine analysis (see Table 1). The genetic test was sent to the Mayo Clinic Medical Laboratories. The results reported that one copy of the Intron 35 (IVs35), nucleotide c. 4460-8G>A. FBN1 variant was deleted (see Table 2). The interpretation of the results reported that the significance of the variant was uncertain, but likely deleterious. The specific alteration had been reported once previously as a disease-causing mutation in a person with a diagnosis of classic Marfan syndrome. After receiving the genetic results, the patient’s rheumatologist, PCP, cardiologist, and geneticist were educated on the results. The patient was then informed of the results and guidance was given for further follow up and evaluation with the author as part of a multidisciplinary approach.
JA was extremely thankful and appreciative of the efforts made to find the diagnosis. JA had been dealing with cardiovascular issues for a number of years with no answers about what was causing them. A few months later JA had the same genetic testing done on his two children. Those results revealed the same genetic mutation that JA had.
Discussion
Ectopia lentis is a displaced or mal-positioned intraocular lens. Ectopia lentis can cause different visual problems for the patient which may include myopia, astigmatism or a loss of accommodative power. Ectopia lentis can be familial or secondary to eye disease and trauma. Trauma is the most common cause of ectopia lentis and usually presents with other signs of ocular trauma.1 Other possible causes of ectopia lentis include cataracts, pseudoexfoliation of the lens capsule, and ciliary body tumors,1 all of which were not seen in the patient. Ciliary body tumors can be seen as elevated lesions in the peripheral fundus. Due to the normal fundus examinations of both eyes and the binocular presentation of the ectopia lentis, a cilary body tumor was not considered in patient JA. Systemic diseases that can cause poor development of the zonules can also cause ectopia lentis. These possible diseases include homocystinuria, Weill-Marchesani syndrome, sulfite oxidase deficiency, and Marfan syndrome.
Homocystinuria is a metabolic disorder that is characterized by increased blood and urine concentrations of homocysteine - a sulfur containing amino acid.11,12 Homocysteine and methionine accumulate in tissues and interfere with collagen cross-linking.13 Skeletal features can include normal to tall stature, fine brittle hair, hypopigmentation, crowded teeth, limited joint mobility and pectus excavatum.13 Ocular features can include dislocation of the lens (usually downward), myopia and glaucoma.11,13 The patient can have mental retardation, seizures and CVAs.11,13 Complications of the disease can include thromboembolism, carotid artery disease, mitral valve prolapse, osteoporosis, fatty infiltration of the liver and pancreatitis.11,13
Weill-Marchesani Syndrome (WMS) is another rare connective tissue disorder. Physical features may include short stature, short and stubby hands and feet, stiff joints, heart defects and sometimes mental retardation.14 Ocular features of the syndrome can include microspherophakia, high myopia, ectopia lentis and glaucoma.14 Ectopia lentis in WMS usually occurs in the second or third decade of life and secondary glaucoma may occur at that time as well.14 WMS patients have weaker than normal lens zonules that can rupture leading to the ectopia lentis. The lens is usually dislocated downward in WMS.1
Sulfite Oxidase Deficiency is a devastating neurologic disease that usually presents in early infancy with seizures and alterations in muscle tone.15 It can result in severe developmental delays and death.15 Sulfite oxidase oxidizes toxic sulfites to non-toxic sulfates in the sulfur amino acid metabolism pathway.15 Ectopia lentis can be a complication of the deficiency. The exact pathophysiology of ectopia lentis in sulfite oxidase deficiency in unknown, but it has been suggested that an increase in matrix metalloproteinases activity might result in increased fibrillin degradation and hence dislocation of the lens.15
Marfan syndrome is one of the most common inherited connective tissue disorders with a reported incidence of between 1/5000 and 1/20,000 depending on the source.2–5,7 Marfan syndrome can affect the ocular, cardiovascular and skeletal systems. The main features of the autosomal dominant disorder include long bone overgrowth, ectopia lentis and aortic root aneurysm.2,3,5,6
The pathogenesis of Marfan syndrome results from an inherited defect in the extracellular glycoprotein called fibrillin-1.2–10 Fibrillin-1 is a large glycoprotein that is thought to provide the structure of extracellular microfibrils that contribute to the formation of elastin and elastic fibers.3,5,7,14 These microfibrils are located throughout the body, but are found in great quantities in the aorta ligaments and ciliary zonules. Analysis of the mutations has found more than 600 mutations of the fibrillin-1 gene on chromosome 15q12 in people with Marfan syndrome.3,16 Most of these mutations cause issues with fibrillin-1.3–5,7 While the abnormal fibrillin-1 can explain the ocular and aorta effects, other factors of Marfan syndrome, such as bone overgrowth, cannot be explained by the changes in tissue elasticity. There is recent evidence that suggests that with the loss of microfibrils, excessive activation of transferring growth factor β (TGF-β) occurs because normal microfibrils seize the TGF- β and control is availability.3,5 When excessive TGF-β is signaled, vascular smooth muscle development, and the strength of the extracellular matrix, can be negatively affected.3,5
People with Marfan syndrome are normally tall with very long extremities and long tapering toes and fingers.3,10 There is a great deal of laxity in the joint ligaments allowing the thumb to be hyper-extended back to the wrist.3,10 The head has bossing frontal eminences and prominent supraorbital ridges.3 Spinal deformities are common including kyphosis and scoliosis.3,10 The chest has a classic deformation, usually pectus excavatum or pigeon-breast deformity.3 The ocular changes that can occur with Marfan syndrome are bilateral ectopia lentis and increased axial length of the globe.3,10 Cardiovascular lesions are the most concerning features of the syndrome. The most common cardiovascular complication is mitral valve prolapse due to the poor structure of the valve.3,10 Cystic medionecrosis is a less common cardiovascular complication than mitral valve prolapse but is more of a concern. The cystic medionecrosis can lead to an aortic dissection and aneurysm, causing death.3,10
The diagnosis of Marfan syndrome is based on the revised Ghent nosology of Marfan syndrome. A person who has an aortic root dilatation and ectopia lentis has met enough criteria to be diagnosed with Marfan syndrome.6 A person who has an aortic root dilatation or dissection and identification of a FBN1 mutation, with or without ectopia lentis, is enough to diagnose Marfan syndrome.6 If a person has an aortic root dilatation or dissection and a FBN1 status that is either normal or unknown, a Marfan syndrome diagnosis can be made if the person has enough other systemic signs even if the patient does not present with ectopia lentis.6 If a person is found to have ectopia lentis but does not have an aortic root dilatation or dissection, a Marfan syndrome diagnosis can be made only if a FBN1 mutation is found that has been associated with aortic problems in the past.6 The genetic report stated that the FBN1 gene mutation had been previously reported one other time in a patient with a classic presentation of Marfan syndrome including cardiovascular involvement. This report, along with the presence of ectopia lentis, confirmed the patient’s diagnosis of Marfan syndrome.
The care of a patient with Marfan syndrome needs to be properly managed, with echocardiograms one or more times per year.2,6 Because aortic root dilatation is usually progressive, the absence of aortic root enlargement on initial clinical examination does not necessarily mean that it will not occur.6 More frequent imaging is recommended when the aortic diameter reaches 45mm or shows a sudden change.2,6 The most common treatment for the prevention of aortic complications is a β-blockade.6 Elective prophylactic repair of the aorta is preferred before enlargement to 60 mm occurs.2,6 Patients who undergo elective repair, as opposed to emergent repair, have considerably better outcomes.2 Generally, patients with Marfan syndrome should avoid contact sports, exercising to the point of exhaustion, and activities that include a valsalva maneuver.6 Yearly ocular examinations should be performed to monitor for ectopia lentis, cataracts, glaucoma or retinal detachments.6 When patients with Marfan syndrome develop ectopia lentis, annual dilated exams should be done to monitor for changes to the subluxation and to watch for other ocular complications. In younger patients that are diagnosed with ectopia lentis for the first time, a dilated exam at six months may be warranted. Children with Marfan syndrome should be monitored closely for refractive changes that could lead to amblyopia. Reasons to consider surgical lens extraction in a Marfan patient include a visually significant cataract, anisometropia, impending complete luxation and lens-induced glaucoma or iritis.6
Conclusion
This case demonstrates the importance of a thorough, comprehensive eye examination. It also demonstrates the importance of a thorough evaluation of a patient who presents with ocular signs that may be manifestations of a systemic condition. Because Marfan syndrome has morbidity risk, anyone found to have ectopia lentis should have a thorough work-up to determine any possible underlying disorder, which should include a cardiovascular exam.