

Introduction
Macular telangiectasia is usually divided into three main types. Type 1 is congenital, typically unilateral, believed to be a variant of Coats disease, and is relatively uncommon. Type 3 is extremely rare, poorly understood, and believed to be an occlusive disease. Type 2 is the most common and the topic of this report. Macular telangiectasia (MacTel) type 2 is an idiopathic bilateral neurodegenerative disease with characteristic changes in the macular capillary network including neurosensory atrophy.1–3 The diagnosis of MacTel is often delayed at the early stages due to lack of clinically noticeable changes, and at later stages, is often misdiagnosed as age-related macular degeneration or other retinal disorders, especially in the presence of neovascularization.1,4 Once patients become symptomatic, it is typically late in the disease process, when there are clinically apparent changes within the macula. Newer imaging modalities have allowed for earlier detection of MacTel by identifying disease specific findings and have become important diagnostic tools.
Case History
A 75-year-old white male presented to the eye clinic for a comprehensive eye exam with complaints of blurred vision, gradually worsening through his habitual glasses at all distances for many years. Relevant ocular history included longstanding bilateral foveal pseudocysts, bilateral refractile retinal particles, and right upper eyelid ptosis and corneal pannus secondary to trauma over 50 years prior. The patient’s medical history was remarkable for systemic hypertension and urinary retention, for which he was taking amlodipine and tamsulosin respectively. The patient had no significant family ocular history and had no history of substance abuse.
Ophthalmic examination revealed stable best corrected visual acuities (BCVA) of 20/40 right eye (OD) and 20/50 left eye (OS). All other entrance testing and anterior segment findings were normal except for the previously noted right upper eyelid ptosis and corneal pannus OD. Intraocular pressures were normal and symmetric.
_and_os_(right_image).png)
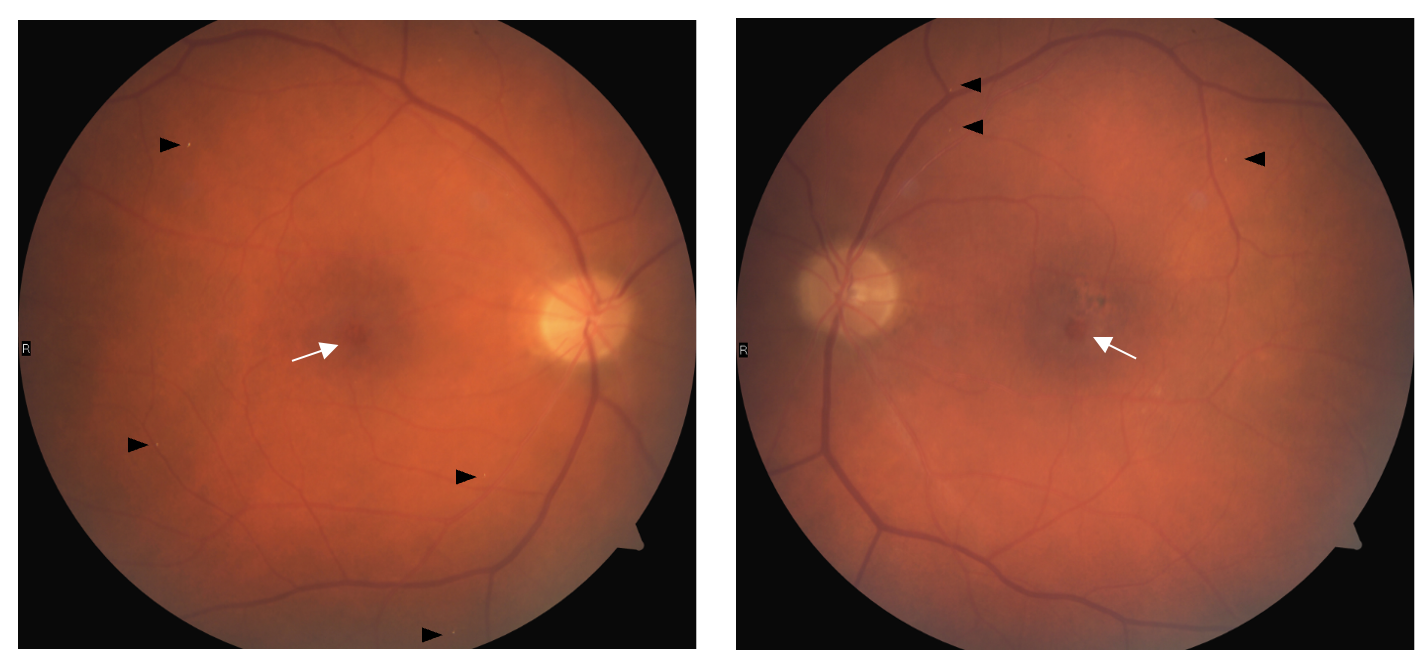
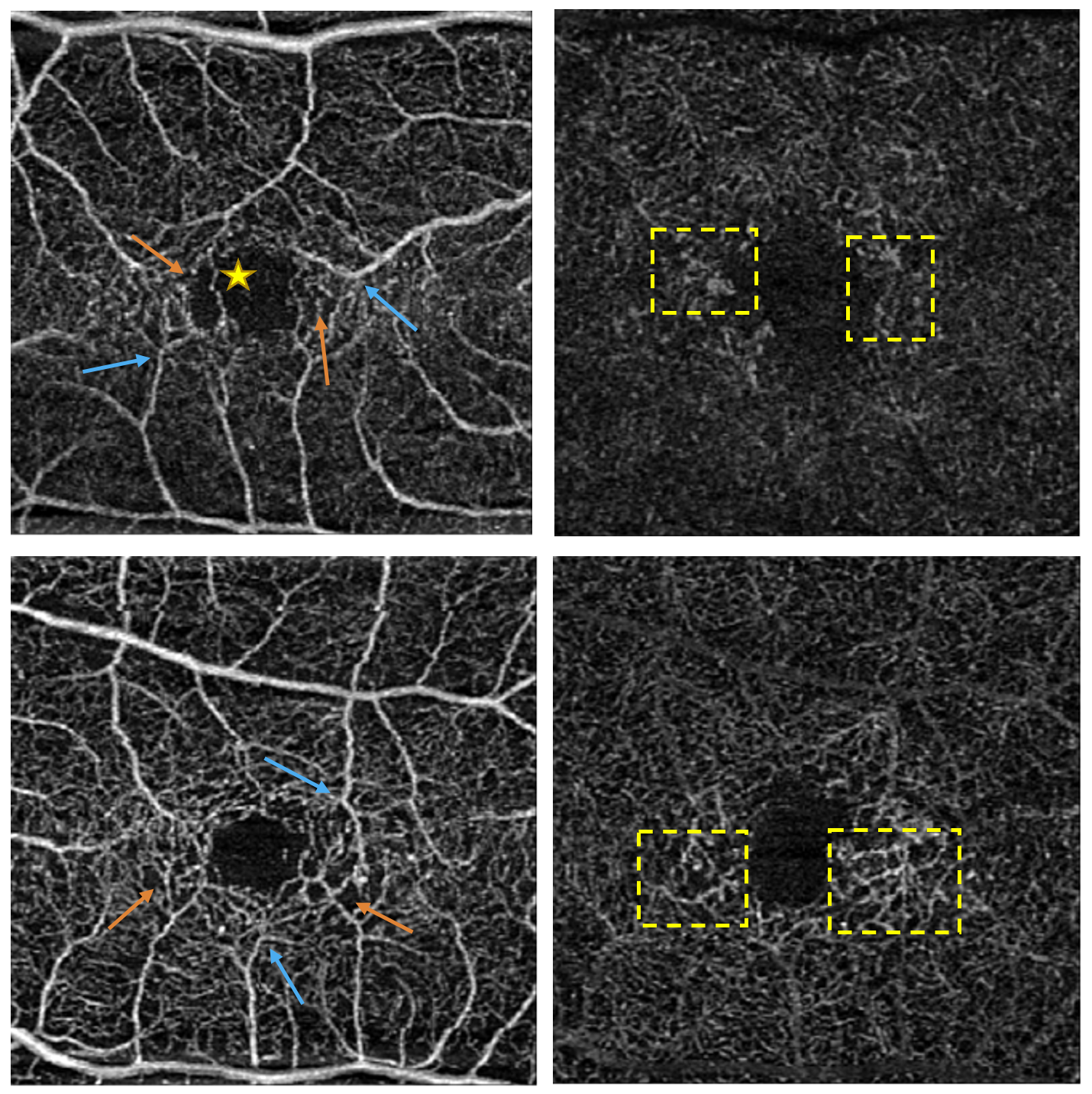
Dilated examination revealed trace nuclear sclerosis and 1+ anterior cortical spoking outside the visual axis OD/OS. Fundus examination revealed optic nerves with distinct margins and intact rim tissue with cup-to-disc ratios assessed as 0.20 round OD and 0.25 round OS. The macula was noted as flat with a central circular red spot in both eyes (OU) with adjacent pigment mottling and clumping superotemporal OS (Figure 1). Multiple refractile deposits were present bilaterally and did not appear to be within the retinal blood vessels (Figure 1). Optical coherence tomography (OCT) of the macula showed internal limiting membrane (ILM) drape, and inner retinal hyporeflective cavity OU with disruption to the ellipsoid zone (EZ) OS (Figure 2). Optical coherence tomography angiography (OCTA) showed diffuse dilated and telangiectatic capillary vessels in both the superficial and deep capillary plexus OU. Focal areas of capillary depletion are also visualized in the superficial capillary plexus with correlated areas of focal microvascular dilatations in the deep capillary plexus OU. Right-angled vessels are also visible in the superficial capillary plexus OU with an anomalous capillary crossing the foveal avascular zone OD (Figure 3).
_and_os_(right_image).png)
_and_deep_capillary_plexu.png)
Based on clinical findings as well as OCT and OCTA imaging results, the diagnosis of MacTel Type 2 was confirmed. The patient’s condition remained in the non-proliferative phase and his visual acuity remained stable for six years. The patient was educated on the condition as well as prognosis. A follow up was recommended in 6 months with repeat OCT and OCTA imaging to continue monitoring for progression and the development of proliferative changes.
Epidemiology
The prevalence of MacTel type 2 has been analyzed in four population-based studies, including two populations of mainly White participants from the United States and Australia, and two others of Black participants from Kenya and Nigeria. The prevalence ranged from 0.005% to 0.1%,5–7 but given the grading being based on color fundus images, without the use of more advanced imaging technologies, the true prevalence is likely to be underestimated. More advanced imaging technologies, including OCT, have been shown to be more sensitive in detecting early and asymptomatic disease stages of MacTel type 2.8
Signs and Symptoms
Patients with MacTel Type 2 often report initial symptoms of metamorphopsia, difficulty reading, or a paracentral scotoma, with the highest incidence of initial symptoms occurring in the fifth to seventh decade of life, although the age of onset can vary significantly.2,6,9 BCVA is typically well preserved over prolonged periods of time with progressive BCVA loss typically slow with one study reporting a mean decrease of approximately one letter per year with a mean follow-up of 4.2 years.10 A 2018 cross-sectional multicenter study evaluated frequency distribution of BCVA in patients with MacTel Type 2. The authors found in their large cohort (4449 eyes of 2248 patients) that nearly 60% of eyes retained a visual acuity of 20/50 or better, and only a few (0.7%) demonstrated bilateral acuity loss of 20/200 or worse.9
By definition, MacTel Type 2 is bilateral, although the presentation may be asymmetric with subtle fundoscopic findings that may be difficult to visualize in the seemingly unaffected eye. Retinal changes typically begin in the temporal paracentral area and may progress to affect a characteristic horizontally elongated oval region approximately one disc diameter eccentricity from the foveal center.1 The first visible fundoscopic change typical for MacTel Type 2 is a reduction in retinal transparency or “retinal graying” in the parafoveal area. The exact cause of this loss of retinal transparency is unknown at this time but is thought to be related to Müller cell dysfunction and/or structural changes within the neurosensory retina. At later disease stages, with progressing neurosensory atrophy, this finding may not be visible.1
Another finding that may be present in early disease as well as all disease stages are crystalline deposits. These deposits appear highly reflective, white, or golden yellow and are located at the inner surface of the retina, within the nerve fiber layer.1,11 The distribution pattern is typically annular within an approximately 4000 micrometer (µm) diameter circular area centered on the fovea and has a strong association with loss of retinal transparency, loss of macular pigment, and fluorescein leakage.11 The exact physical and chemical properties of crystalline deposits associated with MacTel Type 2 is not known at this time. But based on location within the retina, size, distribution pattern, and reflective properties, it is hypothesized they may be composed of retinoids originating in the visual cycle and located within Müller cells that surround the nerve fibers.11 The patient in this case report did present with bilateral crystalline deposits, although the distribution of the visible deposits was generally more peripheral. In general, more deposits are likely to be present in the retina than those detectable in a single fundus image.
Retinal vascular abnormalities, although difficult to visualize on fundus examination, become more apparent as the disease progresses. Both ectasia of retinal capillaries and blunted, dilated venules can occur in isolation or concurrent. These findings can affect both the superficial and deep capillary network and will be discussed in further detail in the diagnosis and imaging section.
Retinal pigment plaques or pigment hyperplasia is another clinical finding suggestive of MacTel Type 2. It is often preceded by atrophic changes in the photoreceptor layer which tends to promote pigment hyperplasia and migration into the neurosensory retina and is often associated with blunted tips of dilated venules. Given the correlation with dilated venules, the suspected pathophysiology is similar to bone spicule-like pigmentation in retinitis pigmentosa where photoreceptor atrophy results in direct contact between retinal vessels and the retinal pigment epithelium leading to pigment migration around retinal vessels.1,9
Foveal atrophy can occur presenting broadly depending on the severity of disease. Early in the disease process foveal reflectivity can be altered or with further progression a pseudo-lamellar hole with cystic formations can be present, like the patient in this case report. As degeneration of the neurosensory retina progresses, true lamellar holes or even full thickness macular holes (FTMH) can occur and may show a small temporal displacement from the foveal center.1 A FTMH secondary to MacTel Type 2 is generally considered a poor surgical candidate due to the neurodegenerative etiology of the hole formation and poor stability of the foveal structure.1,3
Another potential vision threatening complication is the development of a neovascular complex. These complexes can develop at any point in the disease process and seem to originate in the retinal vasculature rather than the choroidal vasculature as seen most of the time in age-related macular degeneration. With progressing disease, these neovascular complexes can infiltrate the subretinal space and develop choroid-retinal shunts.1 Typical clinical findings in the presence of neovascular complexes are intra- and/or sub-retinal edema, intra- and/or sub-retinal hemorrhages, and yellow exudates.
In 1993, Donald Gass and Barbara Blodi suggested a five-stage classification system for MacTel Type 2 based solely on retrospective analysis of fundus photography and fluorescein angiographic findings.12 The authors suggested a temporal sequence of stages that appears to be in line with clinical experience, and despite its longevity, remains the best staging system to date. The main limitation to using this staging system is that it does not consider disease specific characteristics that have become apparent with novel imaging techniques such as OCT, OCTA, fundus autofluorescence, or microperimetry. More research is needed to determine a more accurate and updated staging system that includes these types of imaging modalities.
Differential Diagnoses
There are many ocular and systemic diseases that may manifest similarly to MacTel Type 2. Rare inherited dystrophies may present with similar neurosensory atrophy, or Sjogren-Larsson Syndrome may present with neurosensory atrophy, loss of macular pigment, and superficial crystalline deposits, but this discussion will be limited to more commonly encountered conditions.
Retinal vascular conditions such as diabetic retinopathy, retinal vein occlusion, radiation retinopathy, or inflammatory ocular disease can present with telangiectatic vascular changes, specifically in the macular capillary network. A thorough medical history is needed in all cases. Diabetic retinopathy, most retinal vein occlusions, as well as radiation retinopathy typically involve a more extensive retinal area, outside the macular area, and include retinal hemorrhages and soft exudates. Small macular branch retinal vein occlusions would similarly be limited to the macular area but would respect the horizontal raphe and occur distal to an arteriolar-venular crossing. In cases of inflammatory ocular disease, other related findings such as vitreous inflammatory cells and/or vascular sheathing may be present.1,3
The list of conditions that manifest as retinal pigment plaques or pigment hyperplasia is long, but age-related macular degeneration is typically the most common. Early in the disease drusen are usually present, but in the absence of drusen, clinically it can be difficult to differentiate with MacTel Type 2. Later stages characterized by geographic atrophy can also be difficult to differentiate with late stage MacTel Type 2. Neovascular complexes are another potential complication of both diseases, but in age-related macular degeneration, they usually originate from the choroidal vasculature as mentioned previously. Ultimately, accurate diagnosis of MacTel Type 2 at any stage with clinical evaluation alone can be difficult. Additional imaging techniques are helpful in identifying disease specific abnormalities, differentiating other conditions, and making an accurate diagnosis.
Diagnosis and Imaging
Fundus fluorescein angiography (FFA) has been the gold standard for diagnosis of MacTel Type 2. The classic FFA finding is telangiectatic vessels that leak dye, unrelated to cystic spaces, in the parafoveal area.13 However, with the advent of novel imaging modalities, other retinal changes have become apparent, occur separate from vascular alterations seen on FFA, and likely occur prior.1
OCT and OCTA have proven valuable tools in diagnosing MacTel Type 2. Larger longitudinal studies investigating the natural history on OCT imaging are needed, but smaller case studies have shown consistent characteristic findings. The most common OCT findings are present at the foveal center or temporal to the foveal center and consist of inner retinal hyporeflective cavities with or without ILM drape, outer retinal hyporeflective cavities, EZ disruption, and interdigitation zone (IDZ) disruption.4,14–16 Charbel Issa et al.1 proposed a time course of OCT alterations based on case studies, clinical experience, and correlation with other imaging techniques. The earliest subtle changes may include asymmetric temporal enlargement of the foveal pit with the thinnest sector being temporal, as well as reflectivity alterations of the photoreceptor outer segments. Inner retinal hyporeflective cavities may occur either in the foveal pit or temporal, with associated flattening of the foveal pit. Disruption of the hyper-reflective EZ may also occur early in the disease process but can also be observed in later stages and typically occurs temporal to the foveola. As the disease progresses, outer retinal hyporeflective cavities may form due to neurosensory tissue atrophy potentially leading to apposition of inner retinal layers to the pigment epithelium. With degradation of the photoreceptor layer comes the potential for pigment plaques presenting as hyperreflective lesions that can migrate into the retinal tissue. As the neurosensory retina continues to atrophy and there is reactive proliferation of the pigment epithelium, complete atrophy of the outer retina can occur. Similar to other retinal degenerative conditions, secondary neovascular complexes may form and present as hyperreflective lesions located either sub-pigment epithelium or within the outer neurosensory retinal layers.
OCTA has shown high correlation with FFA in the diagnosis of MacTel Type 2 demonstrating its validity compared to the gold standard.17 There are also certain advantages of OCTA over FFA. Not only can retinal and choroidal microvasculature be imaged noninvasively, without the use of dye, but OCTA can also image the deep capillary plexus, which FFA does not do well.18 The most common finding on OCTA are vascular alterations most commonly seen in the deep capillary plexus, and to a lesser extent, the superficial capillary plexus.13,17,18 In earlier forms of the disease, there can be dilation and telangiectasis of capillaries along with density loss in the deep plexus. Like OCT findings, these changes usually start in the temporal parafoveal area. As the disease progresses, capillary density loss can occur in the superficial plexus temporally, progress to involve the nasal parafoveal area in both the superficial and deep plexus, and in more advanced stages complete involvement of the perifoveal region. Right-angle vessels can also be present in the superficial and deep plexus, typically in the presence of pigment plaques. New vessel proliferation can also be identified with OCTA with multiple studies showing OCTA being superior to FFA in detecting neovascularization related to MacTel Type 2.17,19 These new vessels may present in the outer and/or subretinal spaces and are more commonly associated with areas of prominent capillary loss in the superficial and deep plexus.18
Fundus autofluorescence (FAF) has become more widely available and is proven to be useful as an adjunct imaging modality in monitoring for disease progression as well as early detection of disease. There tends to be wide variability in phenotypic presentation depending on disease stage. Typically, early in the disease process with macular pigment redistribution or loss, there is relative increase in FAF signal in the temporal parafoveal area, commonly in a wedge-shaped formation.20 As the disease progresses there can be mixed or decreased signal depending on structural and vascular changes.
Other multimodal imaging modalities such as confocal reflectance imaging, macular pigment density, adaptive optics, and microperimetry are primarily used in research settings but may become more widely available and prove beneficial clinically in the future.
Treatment and Prognosis
The shift in suspected pathogenesis of MacTel Type 2 from a primarily vascular condition to a neurodegenerative disease has changed the approach toward neuro-protective treatment for the nonproliferative phase of the disease. At this time there are no proven treatments for the nonproliferative form of MacTel Type 2, although a phase 2 randomized sham-controlled clinical trial published in 2019 showed promising results. Participants were randomized 1:1 to the surgical implant of intravitreal sustained delivery of ciliary neurotrophic factor (CNTF) vs sham procedure. The study resulted in the eyes receiving sham treatment had a clinically significant 31% greater progression of neurodegeneration than the CNTF-treated eyes, measured as photoreceptor loss on spectral domain OCT. The treatment also proved beneficial in relation to retinal sensitivity, measured using microperimetry, and reading speed.21 A phase 3 clinical trial was launched and in the recruitment phase, but is currently delayed due to the COVID-19 pandemic.
The current standard of care for the treatment of the proliferative phase of MacTel Type 2 is anti-VEGF therapy. While further studies are needed to compare specific anti-VEGF agents and treatment schedules, most studies have shown long-term functional and structural efficacy with use of intravitreal bevacizumab, ranibizumab, and aflibercept.2
A recent large, cross-sectional multicenter study of 4449 eyes in 2248 patients, confirmed that severe vision loss tends to be rare in MacTel Type 2 (Figure 4).9
_of_all_eyes_and_bilate.png)
Somewhat surprisingly, only a mild effect of age was found in this study increasing the risk of severe visual impairment (BCVA, 20/200 or worse) from 3.8% to 5.7% over 10 years.9 If MacTel Type 2 is a progressive neurodegenerative condition, one would expect the number of patients with severe disease to increase more with increasing age. Although specific limitations in this study might be the cause of this, in general, the likelihood of severe vision loss tends to be lower than compared to age-related macular degeneration.
Conclusion
MacTel Type 2 is often diagnosed at later stages of disease when clinical findings become more apparent, and the patient becomes symptomatic. The condition shares certain clinical findings with other degenerative conditions of the macula, and with its onset later in life, is often mis-diagnosed as age-related macular degeneration. With the advent of newer imaging modalities, certain disease specific characteristics have been identified and MacTel Type 2 can be diagnosed at earlier stages. Eye care providers play a role in differentiating and properly diagnosing this condition, monitoring for potential complications, and accurately educating the patient on prognosis, especially given the lower likelihood of severe vision loss in relation to age-related macular degeneration.
Funding
The author has no financial or proprietary interest in any material or method mentioned in this article. This article has been peer reviewed.